
I don’t want to turn my blog writing into a way of promoting my own papers, but a paper we published this week in Phys. Rev. B[1] contains some very useful points for discussion, particularly relating to comparison to experiment (Chapter 18) and the accuracy of DFT (Chapter 9 and this post and this post). An experimental group in UCL were performing STM on bismuth doped Si(001) surfaces, and found some rather unusual images (an illustration is given below). I’ve worked on Si(001) with experimentalists since the start of my doctorate, and had never seen anything like this before.
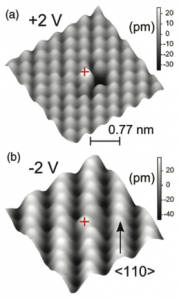
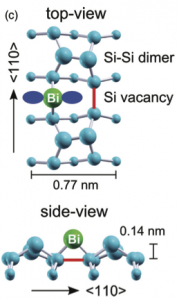
We followed the usual procedures: think about reasonable structures that might be found when imaging Bi-doped Si samples; relax those structures; generate total energies and STM images; compare to experiment. However, the obvious structures (based around substituting a Si atom with a Bi atom) gave simulated STM images which were nothing like the experimental images. My patient experimental co-author had suggested trying a single Bi atom with a missing Si atom (in place of two silicon atoms), but I rejected this on the grounds that it just wouldn’t form. Eventually I tried it, since I’d run out of ideas that I thought were sensible, and found that it matched the STM images perfectly (illustrated below).
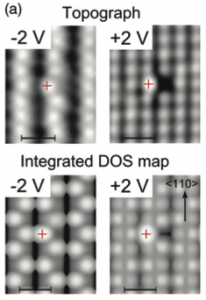
This structure, known as Bi-V, is really very interesting: the Bi has a half-filled, localized orbital which carries an unpaired spin. This is completely different to the bulk behaviour of group V dopants, and the surface behaviour of lighter group V dopants, and results from the large splitting between the 6s and 6p electrons in bismuth (relativistic effects). So: we have a structure that matches experiment, and predicts novel, important electronic properties.
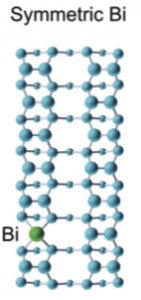
However, the Bi-V structure is found in DFT to be less stable by 0.4eV than a symmetric structure formed by replacing two silicon atoms with the Bi atom located centrally in the hole (images below illustrates Bi-V and symmetric Bi structures). The two structures where Bi replaces a Si dimer are more stable than a Bi-Si heterodimer only if the energy of a dimer vacancy elsewhere on the surface is included (in other words if there is a vacancy on the surface, which is always true, Bi will tend to be found in that vacancy given enough energy to overcome barriers).
This “lack of stability” made several referees unhappy, despite the fact that the sample was prepared in a way that would lead to non-equilibrium structures, and that it’s often possible to have kinetic barriers favouring structures that are not the lowest energy in DFT (see discussion in Section 18.2.1). We subsequently performed hybrid functional (B3LYP) calculations using Gaussian orbitals, and found that the Bi-V structure was more stable than the symmetric structure (though the order depends on the details of the effective core potential used for Bi).
What can we learn from this ? DFT is largely reliable – this is well established – but can be inaccurate. It is possible to fix the inaccuracies, often at some cost, but it can be hard to know what to use. In this case, it seems that the localised state of the Bi was penalised by the DFT self-interaction error, which is remedied by the hybrid functional. But we’re not really sure what percentage of exchange should be mixed in with a hybrid functional, and we know that they can impose other errors (see this previous post).
The bottom line, for me, is that comparison to experiment is more important than absolute DFT energies. But you may have to fight hard to have people believe that.
[1] Phys. Rev. B 88, 035440 DOI:10.1103/PhysRevB.88.035440